








2023版《医疗器械经营质量管理规范》(以下简称“新GSP”)于2024年7月1日正式施行后,GSP审核已全面转向“以主体责任为核心、以风险管控为导向、以全流程追溯为支撑”的新模式。无论是首次认证还是监督审核,企业均需精准把握审核关键内容,规避常见合规风险。本文结合新GSP要求,系统拆解审核核心要点与实操注意事项,为企业高效通过审核提供指引。
一、GSP审核核心内容:聚焦全链条合规
新GSP将审核范围从传统“进销存”延伸至“体系建立、风险防控、新业态管理”等全维度,核心内容可归纳为八大板块,覆盖经营全流程关键节点。
(一)质量管理体系:从“形式合规”到“实质运行”
此为审核首要内容,重点验证体系的完整性与实效性,核心关注:
1. 体系文件适配性:是否结合企业经营品类(如三类植入性器械、体外诊断试剂)及模式(如零售、多仓协同)制定专属文件,避免“照搬模板”;新增的“质量管理体系建立与改进”章节要求是否落实,是否形成“制度-执行-检查-改进”闭环。
2. 主体责任落地:企业负责人每季度质量安全风险会商记录是否完整,是否包含风险识别、措施制定及调度安排;质量负责人职责是否明确,是否具备独立履职权限,三类器械经营企业质量负责人是否存在兼职情况。
3. 持续改进机制:内部审核报告、管理评审报告是否针对前次审核问题及日常风险提出改进措施,整改记录是否包含“原因分析-整改方案-效果验证”全链条资料。
(二)人员管理:资质、能力与健康全覆盖
人员是质量管控的核心载体,审核重点围绕“资质合规、能力匹配、健康达标”展开,差异化要求需精准落地:
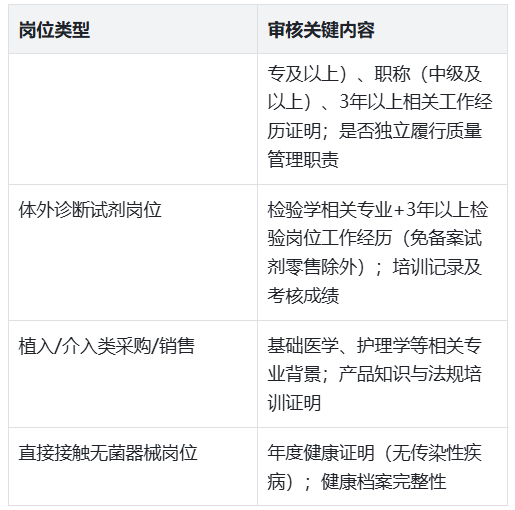
审核组会通过查阅资质文件、现场访谈等方式验证,如随机提问售后人员“不良事件报告流程”,判断培训效果。
(三)设施设备:匹配风险等级与经营规模
硬件设施是质量保障的基础,审核重点为“配置合规、维护到位、运行有效”,核心关注:
1. 仓储设施:功能分区(待验、合格、不合格、发货区)是否物理隔离并标识清晰;冷库/冷藏箱温度是否稳定在2-8℃(针对冷链器械),温度监测设备是否校准并实时上传数据。
2. 专业设备:体外诊断试剂经营企业是否配备离心机、恒温水浴箱等检验设备;无菌器械验收是否配备洁净工作台;自动售械机是否具备温湿度控制与销售记录存储功能。
3. 信息化系统:是否支持UDI全流程追溯(采购、验收、出库等环节);多仓协同企业是否实现跨仓库存统一管控;第三方物流企业是否能通过系统区分自营与受托业务。
(四)采购与验收:源头把控质量关
此环节是防范不合格产品流入的关键,审核重点为“供应商合规、验收规范、记录可溯”:
1. 供应商管理:合格供应商名录是否包含资质审核记录(营业执照、生产/经营许可证、注册证);委托生产品种是否额外收取受托方资料;首营供应商资料是否为加盖公章的复印件或扫描件。
2. 验收操作:验收记录是否包含产品名称、规格、批号、UDI编码等关键信息;无菌器械是否核查灭菌标识与包装完整性;直调产品是否由使用单位与经营企业共同验收并签字确认。

(五)储存与养护:保障产品质量稳定
审核组会通过“现场勘查+记录核查”验证管控效果,核心关注:
1. 储存合规性:货物堆放是否符合“垛间距≥5cm、墙距≥10cm”要求;近效期产品(距有效期6个月内)是否单独标识并优先出库;不合格品是否隔离存放并及时处置。
2. 养护管理:养护计划是否覆盖高风险产品;冷链器械养护记录是否包含每日温湿度数据;养护发现的破损、变质产品是否有闭环处理记录。
(六)销售与售后:确保流向可追溯
审核重点围绕“销售合规、追溯完整、售后响应”展开:
1. 销售管理:销售记录是否与发票、随货同行单信息一致;向非医疗机构销售时是否索取使用需求说明;网络销售是否公示企业与产品资质,交易记录是否同步至信息系统。
2. 售后服务:售后人员是否经考核合格上岗;不良事件报告是否在24小时内上报;产品召回是否及时停止销售并记录召回过程。
(七)UDI与记录管理:全流程追溯落地
新GSP新增重点,审核核心为:
1. UDI实施:三类器械及103种二类器械是否在采购、验收等环节实现UDI关联;集采产品是否按要求“带码招标、采购、结算”。
2. 记录合规:电子记录是否具备防篡改功能,与纸质记录具备同等效力;随货同行单、验收记录等是否至少留存5年;自动售械机销售凭据是否完整。
(八)新业态管理:适配行业发展趋势
针对多仓协同、SPD模式、自动售械机等新业态,审核重点为:
1. 多仓/第三方物流:跨区域仓库是否纳入统一质量管理;第三方物流企业是否通过系统区分不同委托方业务;SPD模式下是否签订质量协议明确双方责任。
2. 自动售械机:是否明确经营主体,设置位置是否合规;内部温湿度是否符合产品要求,定期检查记录是否完整。

二、GSP审核实操注意事项:规避常见合规风险
企业审核失败多源于“细节疏漏”与“文实不符”,结合新GSP要求,需重点关注以下六大注意事项:
(一)避免“文件与现场两张皮”
这是最常见的失分点,如《仓储管理规程》规定“每日巡查”但现场无巡查记录,或SOP要求“UDI扫码验收”但员工仍手工记录。注意事项:审核前开展“文实比对”自查,确保制度要求与现场操作、记录完全一致;重点核对风险会商纪要与实际整改措施的关联性。
(二)人员资质与培训“精准匹配”
审核组会严格核查人员资质的“关联性”与培训的“针对性”。注意事项:体外诊断试剂岗位人员需提供“检验岗位工作经历证明”(而非普通销售经历);植入类产品采购人员需明确其医学相关专业背景(如护理学、医学技术等);培训内容需结合岗位风险,避免“全员统一课件”。
(三)UDI追溯链条“无断点”
UDI管理是新GSP审核必查项,常见问题包括“首营未维护UDI”“销售环节UDI信息缺失”。注意事项:建立UDI专项管理流程,首营时强制索取上游UDI信息并录入系统;严禁供应商与医院间直接借调货,避免UDI追溯断裂;定期核对系统中UDI与产品批号的关联准确性。
(四)冷链管理“全程可控”
冷链器械是高风险审核领域,常见问题为“温度记录不全”“应急预案未演练”。注意事项:冷库温度监测设备需24小时不间断记录,缺失数据需提供合理解释(如设备故障维修证明);应急预案需涵盖断电、设备故障等场景,并每半年至少开展1次包含冷库的实战演练,留存演练照片与总结报告。
(五)直调业务“严守边界”
新GSP收紧直调管理后,违规直调成为重灾区。注意事项:直调仅允许“灾情疫情等特殊情况”或“仅经营大型医用设备”两类场景,需留存特殊情况证明材料;直调首营审核需前置,验收记录需使用单位与经营企业双签字,随货同行单需一式两份分别留存。
(六)电子记录“合规有效”
企业常因电子记录“防篡改功能缺失”“权限管理混乱”失分。注意事项:信息化系统需设置操作日志,记录所有人员的登录、修改行为;电子签章需符合法律要求,与纸质签章具备同等效力;关键记录(如验收记录、销售记录)的电子版本需定期备份,避免数据丢失。
三、审核前核心筹备动作
1. 全流程自查:对照本文审核核心内容,组建跨部门自查小组,重点排查UDI管理、冷链记录、人员资质等薄弱环节,形成自查报告与整改清单。
2. 文件梳理:按“体系文件-操作SOP-记录表单”分类归档,突出新GSP新增要求(如风险会商纪要、UDI管理文件),便于审核组查阅。
3. 人员培训:针对质量负责人、验收、售后等关键岗位,开展新GSP条款专项培训,模拟审核组访谈问题(如“如何处理不良事件”“直调业务流程”),确保回答准确。
4. 现场整改:规范仓储区域标识,清理应急通道障碍物,确保冷链设备正常运行,提前调试信息化系统演示功能(如UDI追溯、多仓库存查询)。
四、结语:以审核为契机构建长效合规机制
新GSP框架下的审核已不再是“一次性考核”,而是推动企业建立长效质量管理机制的重要手段。企业需摒弃“为过审而筹备”的短期思维,将审核关键内容转化为日常经营的管理习惯,通过“人员能力提升、系统功能优化、流程规范落地”,实现“合规达标”与“质量提升”的双重目标,为公众用械安全筑牢防线。









 18576401396
18576401396 



